- The Oligonucleotide Therapeutics Society held its annual meeting this September in Denmark
- ISIS (Click for research page), ALNY (Click for research page), and TKMR (Click for research page) made numerous presentations, including those by Regulus, a joint venture of ISIS and ALNY. I also will note significant presentations by competing company Santaris, the defendant in a recent patent infringement lawsuit filed by Isis.
- Click here for PDF with all available abstracts from the conference.
- Check out info on presentations by BiotechDueDiligence companies at other scientific conferences on the Upcoming Events and Past Events pages.
- Detailed info on the presentations can be found below the jump:
Alnylam Presentations:
Be sure to see the ALNY page for more complete info on the clinical stage programs descirbed below, including ALN-TTR and ALN-VSP02
CHEMICAL STRAT EGIES FOR DELIVERY OF RNAi DRUGS
Muthiah Manoharan
At Alnylam Pharmaceuticals, we have developed and applied multiple chemistry strategies
to address the challenge of cellular delivery of drugs that function through RNAi pathways.
These include chemical modifications of oligonucleotides, molecular conjugates and delivery
systems based on liposomal nanoparticles (LNPs).Our progress in these areas will be summarized. [This abstract lacks details, but I am including it because of the list of relevant references below].
References
I. Chemical Modifications of RNAi
1. Manoharan, M. “RNA interference and chemically modified small interfering RNAs.”
Curr. Opin. Chem. Biol. 2004, 8, 570-579.
2. Bumcrot, D. et al. “RNAi therapeutics: a potential new class of pharmaceutical drugs.”
Nature Chemical Biology 2006, 2, 711-719.
3. Manoharan, M. and Rajeev, K. G. “Utilizing chemistry to harness RNA interference pathways
for therapeutics: chemically modified siRNAs and antagomirs.” Antisense Drug
Technology (2nd Ed.), 2008, 437-464.
4. Zlatev, I. et al. “Efficient solid-phase chemical synthesis of 5’-triphosphates of DNA,
RNA and their analogs.” Organic Letters 2010, 12, 2190-2193.
5. Watts, J. et al. “Effect of chemical modifications on modulation of gene expression by
duplex antigene RNAs that are complementary to non-coding transcripts at gene promoters.”
Nucleic Acids Res. 2010, 38, 5242-5259.
6. Addepalli, H. et al. “Modulation of thermal stability can enhance the potency of siRNA.”
Nucleic Acids Res. 2010, 38, 7320-7331.
7. Manoharan, M. et al. “Unique gene-silencing and structural properties of 2’-F modified
siRNAs.” Angewandte Chemie, (International Edition) 2011, 50, 2284-2288.
8. Pallan, P. S. et al.;. Unexpected origins of the enhanced pairing affinity of 2’-fluoromodifiedRNA. Nucleic Acids Res. 2011, 39, 3482-3495.
II. Conjugates for siRNA and Antagomir Delivery
9. Soutschek, J. et al. “Therapeutic silencing of an endogenous gene by systemic
administration of modified siRNAs.” Nature, 2004, 432, 173-178.
10. Kruetzfeldt, J. et al. “Silencing of microRNAs in vivo with ‘antagomirs’.”
Nature, 2005, 438, 685-689.
11. Wolfrum, C. et al., “Mechanisms and optimization of in vivo delivery of lipophilic
siRNAs.” Nature Biotech. 2007, 25, 1149-1157.
12. Wu, Y. et al. “Durable protection from herpes simplex virus-2 transmission following
intravaginal application of siRNAs targeting both a viral and host gene.” Cell Host &
Microbe 2009, 5, 84-94.
13. Querbes, W. et al. “Direct CNS delivery of siRNA mediates robust silencing inoligodendrocytes.” Oligonucleotides 2009, 19, 23-30.
14. Chen, Q. et al. “Lipophilic siRNAs mediate efficient gene silencing in oligodenrocytes
with direct CNS delivery.” Journal of Controlled Release 2010, 144: 227-232.
15. DiFiglia, M. et al. “Therapeutic silencing of mutant huntingtin with siRNA attenuates
striatal and cortical neuropathology and behavioral deficits.” PNAS, 2007 104,
17204-17209.
16. Alam, Md. R. et al. Multivalent Cyclic RGD Conjugates for Targeted Delivery of Small
Interfering RNA. Bioconjugate Chemistry, 2011, (in press).
17. Jayaprakash, K. N. et al. “Non-nucleoside building blocks for copper-assisted and copper-
free click chemistry for the efficient synthesis of RNA conjugates.” Organic Letters
2010, 12, 5410-5413.
18. Yamada, T. et al. “Versatile site-specific conjugation of small molecules to siRNAs usingclick chemistry.” Org. Chem., 2011, 76, 1198-1211.
III. Liposomal Nanoparticles (LNPs)
19. Zimmermann, T. et al. “RNAi-mediated gene silencing in non-human primates.” Nature,
2006, 441, 111-114.
20. Akinc, A. et al. “A combinatorial library of lipid-like materials for delivery of RNAi
therapeutics.” Nature Biotech. 2008, 26, 561-569.
21. Frank-Kamenetsky, M. et al. “Therapeutic RNAi targeting PCSK9 acutely lowers plasma
cholesterol in rodents and LDL cholesterol in nonhuman primates.” PNAS, 2008, 105,
11915-11920.
22. Akinc, A. et al., “Development of lipidoid-siRNA formulations for systemic delivery to
the liver.” Molecular Therapy, 2009, 17, 872-879.
23. Love, K. T. et al., “Lipid-like materials for low-dose, in vivo gene silencing.”PNAS 2010,
107, 9915.
24. Akinc, A. et al. “Targeted delivery of RNAi therapeutics.” Molecular Therapy 2010, 18,
1357-1364.
25. Semple, S. et al. “Rational design of cationic lipids for siRNA delivery.”Nature Biotechnology 2010, 28, 172-176.
ALN-TTR, AN RNAi THERAPEUTIC FOR THE TREAT MENT OF
TRANSTHYRETIN AMYLOIDOSIS
Dinah W. Y. Sah1, Qingmin Chen1, Susete Costelha2, Jim Butler1, Shannon Fishman1,
Anthony Rossomando1, Lubomir Nechev1, Maria Joao Saraiva2, Teresa Coelho3, Ole B.
Suhr4, David Adams5, Pierre Lozeron5, Philip Hawkins6, Timothy Mant7, Renta Hutabarat1,
Rick Falzone1, Jeff Cehelsky1, Yaysie Figueroa1, Akshay Vaishnaw1, Jared Gollob1
Transthyretin amyloidosis (ATTR) is a fatal, autosomal dominant, multisystem disease
caused by abnormal extracellular deposits of transthyretin (TTR) amyloid that lead to
familial amyloidotic polyneuropathy (FAP) and/or familial amyloidotic cardiomyopathy
(FAC), depending on the sites of deposition. More than 100 TTR mutations have been
reported, with the vast majority resulting in amyloid pathology. Wild-type TTR also contributes
to pathology and clinical progression. There is a high unmet medical need for new therapies,
with liver transplantation being the only available treatment for a subset of FAP patients.
ALN-TTR is a systemically administered lipid nanoparticle-formulation of a small interfering
RNA (siRNA) targeting wild-type and all mutant forms of TTR. This formulation delivers
the siRNA predominantly to the liver, thereby inhibiting TTR synthesis at the primary site
of production. In transgenic mice expressing the human V30M transgene in a heat shock
transcription factor 1 null background, ALN-TTR01 administration led to robust reduction
of TTR mRNA levels in the liver and TTR protein levels in the circulation, and significant
regression of TTR protein in tissues known to be affected by ATTR, including the peripheral
nervous system and gut. These results demonstrate the potential therapeutic benefit of ALNTTR01 for the treatment of ATTR. A Phase 1 randomized, single-blind, placebo-controlled
clinical trial of ALN-TTR01 is underway in Portugal, Sweden, the United Kingdom and
France. The primary objective is to evaluate the safety and tolerability of a single intravenous
dose of ALN-TTR01 in patients with ATTR. Secondary objectives include characterization
of plasma and urine drug pharmacokinetics, and assessment of pharmacodynamic activity
based on measurements of circulating TTR serum levels. In this presentation, an update on
the clinical development of ALN-TTR01 will be provided, as well as an update on the status of a second generation RNAi therapeutic targeting TTR.
EPATOCYTE SPECIFIC TARGETING AND DELIVERY OF siRNACARBOHYDRATE
CONJUGATES
Kallanthottathil G. Rajeev, K. NarayanannairJayaprakash, Gang Wang, Ligang Zhang, Chang
GengPeng, Jennifer Sherman, Mariano Severgnini, Amy Chan, Klaus Charisse,
Renta Hutabarat, Martin Maier, Kevin Fitzgerald,Dinah W.Y. Sah, Muthiah Manoharan,
Kenneth Koblan and Tracy S. Zimmermann
The asialoglycoprotein receptor (ASGPR) is a transmembrane receptor that mediates clearance
of extracellular glycoproteins with exposed terminal galactose residues. Thisreceptor
is highly expressed on the surface of liver hepatocytes and facilitates multiple rounds of
uptake and clearance of glycoproteins. These features of the ASGPR along with its ligand
specificity make it an attractive strategy for liver-specific delivery of galactosylated-drugs.
We have shown that conjugation of a triantennary N-acetylgalactosamine ligand to a siRNA
(siRNA-GalNAc) has nM binding affinity to the ASGPR and facilitates uptake and gene
silencing both in vitro and in vivo. We will describe further improvements ondelivery of
siRNA-GalNAc conjugates that lead to enhanced siRNA pharmacology. The results to
be presented include the impact of mode of administration on efficacy, application across
multiple targets in hepatocytes, and demonstration of liver-specific gene silencing at low mg/kg siRNA-GalNAc doses in mouse, rat and non-human primate.
MECHANISTIC INSIGHTS INTO LIPID NANOPARTICLE-MEDIATED
DELIVERY OF siRNA
Martin A. Maier1, Jerome Gilleron3, William Querbes1, Xuemei Zhang1, Valerie Clausen1,
Renta Hutabarat1, Yosef Landesman1, Nenad Svrzikapa1, June Qin1, Undine Schubert3,
Muthusamy Jayaraman1, Michael J. Hope2, Dinah W. Y. Sah1, Kevin Fitzgerald1,
Kallanthottathil G. Rajeev1, Mark A. Tracy1, Rachel Meyers1, Muthiah Manoharan1,
Marino Zerial3, Akin Akinc1
Safe and efficient delivery of short interfering RNAs (siRNAs) is a prerequisite for the development and advancement of RNAi therapeutics as a new class of innovative medicines. Lipid nanoparticles (LNPs) are among the most efficient carriers for systemic delivery of siRNA
and have been successfully employed to silence therapeutically relevant genes in a number
of species from rodents to non-human primates. In recent years, multiple liver-targeted
RNAi programs utilizing LNP-formulated siRNAs have advanced into human clinical trials.
Meanwhile, the discovery of novel lipid materials and the recent progress on the mechanistic
understanding of the LNP platform has enabled the development of the next generation LNPs
with improved therapeutic indices. Comparing two distinct classes of lipid-based nanoparticles,
we have previously shown that apolipoprotein E acts as an endogenous targeting ligand
for ionizable lipid nanoparticles but not for cationic lipid nanoparticles, which carry a permanent
positive charge at physiologic pH. At the cellular level, functional delivery requires the
release of siRNA from the endosomal compartment into the cytoplasm. Electron microscopy
(EM) was used to track the cellular uptake and intracellular trafficking of siRNA labeled with
gold nanoparticles and formulated in LNPs. Based on EM analysis of cultured cells as well as
liver sections from treated animals, we estimate that a low percentage of the Au-labeled siRNA
escapes the endosomal pathway and reaches the cytosol, which is in good agreement with
time course studies using stem-loop PCR for siRNA quantification. These results help provide
insights into the intracellular mechanism of LNP-mediated siRNA delivery, which may facilitatefurther rational optimization of this siRNA delivery platform.
ACTIVATION OF CYCLOOXYGENASE-2 (COX-2) GENE
EXPRESSION BY PROMOTER-TARGETED DUPLEX RNAs
Masayuki Matsui1, Huiying Zhang1, Yougjun Chu1, Klaus Charisse2, Muthiah Manoharan2,
David R. Corey1, and Bethany A. Janowski1
RNA-mediated gene activation has been investigated over several years in some genes such
as progesterone receptor (PR), LDL receptor (LDLR), p21, and E-cadherin, but its mechanism
is not fully understood yet. To obtain more insights into mechanism of RNA-mediated
gene activation, we targeted cyclooxygenase-2 (COX-2) gene using promoter-targeted duplex
RNAs (antigene RNAs (agRNA)). COX-2 is a rate-limiting enzyme responsible for prostaglandin
biosynthesis, and COX-2 gene expression is associated with tumorigenesis, inflammation,
and apoptosis. Small RNAs that modulate COX-2 gene expression would be useful
as tools to investigate biological functions of COX-2 in cells.
To investigate whether agRNAs have the potential to modulate COX-2 gene expression, duplex
RNAs targeting the promoter were designed and tested in A549 lung cancer cells for
gene activation. After transfecting agRNAs, western blot and qPCR analysis were performed
to check expression levels of COX-2 protein and mRNA. We found that some of the duplex
RNAs (agRNA-12 etc; 25 nM) can activate COX-2 gene expression by more than 20-fold
relative to mismatch controls. The fold activation was similar with that for IL-1b (10 ng/mL),
a natural activator for COX-2 gene. We observed more than 100-fold activation in COX-2
mRNA in cells treated with both the activating agRNA and IL-1b, which indicates the two
activators can work synergistically. ChIP analysis showed increased occupancy of RNA polymerase II by 3-5 fold and NFkB by 2-4 fold in the promoter, suggesting that this activation
occurs at the level of transcription. In our previous studies with PR and LDLR, we identified
non-coding transcripts overlapping their promoters, which could be potential targets for agRNAs.
We performed RACE and strand-specific RT-qPCR to examine whether similar noncoding
transcripts are expressed in the COX-2 gene promoter. Those analyses revealed that
sense and antinsense transcripts are expressed at the COX-2 gene promoter. These transcripts
might be an endogenous mediator for regulating COX-2 gene expression that can be targeted
by agRNAs. Transfection of activating agRNAs did not substantially change expression of interferon responsive genes such as OAS1, OAS2, MX1, IFITM1, and ISGF3g, suggesting that
the activation of COX-2 gene by agRNAs is not due to interferon response. These data suggest
that COX-2 gene can be induced by promoter-targeted duplex RNAs and the gene can beused as a model for research of RNA-mediated gene activation.
RNAi in humans: Phase I dose-escalation study of ALN-VSP02, a novel RNAi therapeutic for solid tumors with liver involvement
Background: ALN-VSP02 is a RNA interference (RNAi) therapeutic comprised of lipid
nanoparticle-formulated small interfering RNAs (siRNAs) targeting the expression of vascular
endothelial growth factor (VEGF)-A and kinesin spindle protein (KSP).
Methods: A multi-center, open label, Phase 1 dose-escalation trial of ALN-VSP02 administered
as a 15-minute iv infusion q2 wks was initiated in patients (pts) with advanced solid
tumors and at least one measurable liver lesion. Main objectives included evaluation of
safety/tolerability and assessment of PK/PD.
Results: Forty-one pts were enrolled across 7 dose levels (0.1-1.5 mg/kg); median age 57 yrs,
all with multiple prior therapies. A total of 182 doses have been administered to date, mean of
4.4 (range 1-24). Treatment was generally well-tolerated; the most common AEs were lowgrade
nausea, fatigue and fever with no clear dose-dependence. One on-study death (liver
failure in a pt with near complete replacement of the liver by tumor) deemed possibly related
to treatment occurred at 0.7 mg/kg. Grade 2 infusion-related reactions occurred in 15% of
pts or 3% of total doses administered and were managed with slowing of infusion. Grade 1-2
chills/rigors were seen in 3 of 11 pts at 1.25 mg/kg. Dose-limiting toxicities included reversible
grade 3 thrombocytopenia (2 pts at1.25 mg/kg) and hypokalemia (1 pt at 1.5 mg/kg).
Plasma PK showed dose-proportional AUC and Cmax. Post-treatment biopsies from 10 pts (7
liver and 3 extrahepatic tumors) showed pharmacologically relevant concentrations (2.2-142
ng/g tissue) of both siRNAs. Molecular evidence of RNAi-mediated VEGF mRNA cleavage
was shown in liver tumor biopsies (n=2 at 0.4 mg/kg) and in an extrahepatic tumor biopsy
(ovarian cancer at 1.25 mg/kg) through use of the 5’ RACE assay. Additional evidence for
an anti-VEGF effect with ALN-VSP02 included a decrease in Ktrans of at least 40% by
DCE-MRI in 46% of evaluable pts. Among 37 pts evaluable for response, 8% (1 of 13) at
doses ≤ 0.4 mg/kg had stable disease (SD) for at least 2 mo compared to 50% (12/24) with
SD (n=11) or PR (n=1, endometrial cancer with liver metastases) at doses ≥ 0.7 mg/kg.
Conclusions: ALN-VSP02 is well-tolerated and has antitumor activity. Pharmacodynamic
data are consistent with an anti-VEGF effect, and 1.0 mg/kg q2wks is the recommendedPhase II dose.
Be sure to see the ALNY page for more complete info on the clinical stage programs descirbed below, including ALN-TTR and ALN-VSP02
CHEMICAL STRAT EGIES FOR DELIVERY OF RNAi DRUGS
Muthiah Manoharan
At Alnylam Pharmaceuticals, we have developed and applied multiple chemistry strategies
to address the challenge of cellular delivery of drugs that function through RNAi pathways.
These include chemical modifications of oligonucleotides, molecular conjugates and delivery
systems based on liposomal nanoparticles (LNPs).Our progress in these areas will be summarized. [This abstract lacks details, but I am including it because of the list of relevant references below].
References
I. Chemical Modifications of RNAi
1. Manoharan, M. “RNA interference and chemically modified small interfering RNAs.”
Curr. Opin. Chem. Biol. 2004, 8, 570-579.
2. Bumcrot, D. et al. “RNAi therapeutics: a potential new class of pharmaceutical drugs.”
Nature Chemical Biology 2006, 2, 711-719.
3. Manoharan, M. and Rajeev, K. G. “Utilizing chemistry to harness RNA interference pathways
for therapeutics: chemically modified siRNAs and antagomirs.” Antisense Drug
Technology (2nd Ed.), 2008, 437-464.
4. Zlatev, I. et al. “Efficient solid-phase chemical synthesis of 5’-triphosphates of DNA,
RNA and their analogs.” Organic Letters 2010, 12, 2190-2193.
5. Watts, J. et al. “Effect of chemical modifications on modulation of gene expression by
duplex antigene RNAs that are complementary to non-coding transcripts at gene promoters.”
Nucleic Acids Res. 2010, 38, 5242-5259.
6. Addepalli, H. et al. “Modulation of thermal stability can enhance the potency of siRNA.”
Nucleic Acids Res. 2010, 38, 7320-7331.
7. Manoharan, M. et al. “Unique gene-silencing and structural properties of 2’-F modified
siRNAs.” Angewandte Chemie, (International Edition) 2011, 50, 2284-2288.
8. Pallan, P. S. et al.;. Unexpected origins of the enhanced pairing affinity of 2’-fluoromodifiedRNA. Nucleic Acids Res. 2011, 39, 3482-3495.
II. Conjugates for siRNA and Antagomir Delivery
9. Soutschek, J. et al. “Therapeutic silencing of an endogenous gene by systemic
administration of modified siRNAs.” Nature, 2004, 432, 173-178.
10. Kruetzfeldt, J. et al. “Silencing of microRNAs in vivo with ‘antagomirs’.”
Nature, 2005, 438, 685-689.
11. Wolfrum, C. et al., “Mechanisms and optimization of in vivo delivery of lipophilic
siRNAs.” Nature Biotech. 2007, 25, 1149-1157.
12. Wu, Y. et al. “Durable protection from herpes simplex virus-2 transmission following
intravaginal application of siRNAs targeting both a viral and host gene.” Cell Host &
Microbe 2009, 5, 84-94.
13. Querbes, W. et al. “Direct CNS delivery of siRNA mediates robust silencing inoligodendrocytes.” Oligonucleotides 2009, 19, 23-30.
14. Chen, Q. et al. “Lipophilic siRNAs mediate efficient gene silencing in oligodenrocytes
with direct CNS delivery.” Journal of Controlled Release 2010, 144: 227-232.
15. DiFiglia, M. et al. “Therapeutic silencing of mutant huntingtin with siRNA attenuates
striatal and cortical neuropathology and behavioral deficits.” PNAS, 2007 104,
17204-17209.
16. Alam, Md. R. et al. Multivalent Cyclic RGD Conjugates for Targeted Delivery of Small
Interfering RNA. Bioconjugate Chemistry, 2011, (in press).
17. Jayaprakash, K. N. et al. “Non-nucleoside building blocks for copper-assisted and copper-
free click chemistry for the efficient synthesis of RNA conjugates.” Organic Letters
2010, 12, 5410-5413.
18. Yamada, T. et al. “Versatile site-specific conjugation of small molecules to siRNAs usingclick chemistry.” Org. Chem., 2011, 76, 1198-1211.
III. Liposomal Nanoparticles (LNPs)
19. Zimmermann, T. et al. “RNAi-mediated gene silencing in non-human primates.” Nature,
2006, 441, 111-114.
20. Akinc, A. et al. “A combinatorial library of lipid-like materials for delivery of RNAi
therapeutics.” Nature Biotech. 2008, 26, 561-569.
21. Frank-Kamenetsky, M. et al. “Therapeutic RNAi targeting PCSK9 acutely lowers plasma
cholesterol in rodents and LDL cholesterol in nonhuman primates.” PNAS, 2008, 105,
11915-11920.
22. Akinc, A. et al., “Development of lipidoid-siRNA formulations for systemic delivery to
the liver.” Molecular Therapy, 2009, 17, 872-879.
23. Love, K. T. et al., “Lipid-like materials for low-dose, in vivo gene silencing.”PNAS 2010,
107, 9915.
24. Akinc, A. et al. “Targeted delivery of RNAi therapeutics.” Molecular Therapy 2010, 18,
1357-1364.
25. Semple, S. et al. “Rational design of cationic lipids for siRNA delivery.”Nature Biotechnology 2010, 28, 172-176.
ALN-TTR, AN RNAi THERAPEUTIC FOR THE TREAT MENT OF
TRANSTHYRETIN AMYLOIDOSIS
Dinah W. Y. Sah1, Qingmin Chen1, Susete Costelha2, Jim Butler1, Shannon Fishman1,
Anthony Rossomando1, Lubomir Nechev1, Maria Joao Saraiva2, Teresa Coelho3, Ole B.
Suhr4, David Adams5, Pierre Lozeron5, Philip Hawkins6, Timothy Mant7, Renta Hutabarat1,
Rick Falzone1, Jeff Cehelsky1, Yaysie Figueroa1, Akshay Vaishnaw1, Jared Gollob1
Transthyretin amyloidosis (ATTR) is a fatal, autosomal dominant, multisystem disease
caused by abnormal extracellular deposits of transthyretin (TTR) amyloid that lead to
familial amyloidotic polyneuropathy (FAP) and/or familial amyloidotic cardiomyopathy
(FAC), depending on the sites of deposition. More than 100 TTR mutations have been
reported, with the vast majority resulting in amyloid pathology. Wild-type TTR also contributes
to pathology and clinical progression. There is a high unmet medical need for new therapies,
with liver transplantation being the only available treatment for a subset of FAP patients.
ALN-TTR is a systemically administered lipid nanoparticle-formulation of a small interfering
RNA (siRNA) targeting wild-type and all mutant forms of TTR. This formulation delivers
the siRNA predominantly to the liver, thereby inhibiting TTR synthesis at the primary site
of production. In transgenic mice expressing the human V30M transgene in a heat shock
transcription factor 1 null background, ALN-TTR01 administration led to robust reduction
of TTR mRNA levels in the liver and TTR protein levels in the circulation, and significant
regression of TTR protein in tissues known to be affected by ATTR, including the peripheral
nervous system and gut. These results demonstrate the potential therapeutic benefit of ALNTTR01 for the treatment of ATTR. A Phase 1 randomized, single-blind, placebo-controlled
clinical trial of ALN-TTR01 is underway in Portugal, Sweden, the United Kingdom and
France. The primary objective is to evaluate the safety and tolerability of a single intravenous
dose of ALN-TTR01 in patients with ATTR. Secondary objectives include characterization
of plasma and urine drug pharmacokinetics, and assessment of pharmacodynamic activity
based on measurements of circulating TTR serum levels. In this presentation, an update on
the clinical development of ALN-TTR01 will be provided, as well as an update on the status of a second generation RNAi therapeutic targeting TTR.
EPATOCYTE SPECIFIC TARGETING AND DELIVERY OF siRNACARBOHYDRATE
CONJUGATES
Kallanthottathil G. Rajeev, K. NarayanannairJayaprakash, Gang Wang, Ligang Zhang, Chang
GengPeng, Jennifer Sherman, Mariano Severgnini, Amy Chan, Klaus Charisse,
Renta Hutabarat, Martin Maier, Kevin Fitzgerald,Dinah W.Y. Sah, Muthiah Manoharan,
Kenneth Koblan and Tracy S. Zimmermann
The asialoglycoprotein receptor (ASGPR) is a transmembrane receptor that mediates clearance
of extracellular glycoproteins with exposed terminal galactose residues. Thisreceptor
is highly expressed on the surface of liver hepatocytes and facilitates multiple rounds of
uptake and clearance of glycoproteins. These features of the ASGPR along with its ligand
specificity make it an attractive strategy for liver-specific delivery of galactosylated-drugs.
We have shown that conjugation of a triantennary N-acetylgalactosamine ligand to a siRNA
(siRNA-GalNAc) has nM binding affinity to the ASGPR and facilitates uptake and gene
silencing both in vitro and in vivo. We will describe further improvements ondelivery of
siRNA-GalNAc conjugates that lead to enhanced siRNA pharmacology. The results to
be presented include the impact of mode of administration on efficacy, application across
multiple targets in hepatocytes, and demonstration of liver-specific gene silencing at low mg/kg siRNA-GalNAc doses in mouse, rat and non-human primate.
MECHANISTIC INSIGHTS INTO LIPID NANOPARTICLE-MEDIATED
DELIVERY OF siRNA
Martin A. Maier1, Jerome Gilleron3, William Querbes1, Xuemei Zhang1, Valerie Clausen1,
Renta Hutabarat1, Yosef Landesman1, Nenad Svrzikapa1, June Qin1, Undine Schubert3,
Muthusamy Jayaraman1, Michael J. Hope2, Dinah W. Y. Sah1, Kevin Fitzgerald1,
Kallanthottathil G. Rajeev1, Mark A. Tracy1, Rachel Meyers1, Muthiah Manoharan1,
Marino Zerial3, Akin Akinc1
Safe and efficient delivery of short interfering RNAs (siRNAs) is a prerequisite for the development and advancement of RNAi therapeutics as a new class of innovative medicines. Lipid nanoparticles (LNPs) are among the most efficient carriers for systemic delivery of siRNA
and have been successfully employed to silence therapeutically relevant genes in a number
of species from rodents to non-human primates. In recent years, multiple liver-targeted
RNAi programs utilizing LNP-formulated siRNAs have advanced into human clinical trials.
Meanwhile, the discovery of novel lipid materials and the recent progress on the mechanistic
understanding of the LNP platform has enabled the development of the next generation LNPs
with improved therapeutic indices. Comparing two distinct classes of lipid-based nanoparticles,
we have previously shown that apolipoprotein E acts as an endogenous targeting ligand
for ionizable lipid nanoparticles but not for cationic lipid nanoparticles, which carry a permanent
positive charge at physiologic pH. At the cellular level, functional delivery requires the
release of siRNA from the endosomal compartment into the cytoplasm. Electron microscopy
(EM) was used to track the cellular uptake and intracellular trafficking of siRNA labeled with
gold nanoparticles and formulated in LNPs. Based on EM analysis of cultured cells as well as
liver sections from treated animals, we estimate that a low percentage of the Au-labeled siRNA
escapes the endosomal pathway and reaches the cytosol, which is in good agreement with
time course studies using stem-loop PCR for siRNA quantification. These results help provide
insights into the intracellular mechanism of LNP-mediated siRNA delivery, which may facilitatefurther rational optimization of this siRNA delivery platform.
ACTIVATION OF CYCLOOXYGENASE-2 (COX-2) GENE
EXPRESSION BY PROMOTER-TARGETED DUPLEX RNAs
Masayuki Matsui1, Huiying Zhang1, Yougjun Chu1, Klaus Charisse2, Muthiah Manoharan2,
David R. Corey1, and Bethany A. Janowski1
RNA-mediated gene activation has been investigated over several years in some genes such
as progesterone receptor (PR), LDL receptor (LDLR), p21, and E-cadherin, but its mechanism
is not fully understood yet. To obtain more insights into mechanism of RNA-mediated
gene activation, we targeted cyclooxygenase-2 (COX-2) gene using promoter-targeted duplex
RNAs (antigene RNAs (agRNA)). COX-2 is a rate-limiting enzyme responsible for prostaglandin
biosynthesis, and COX-2 gene expression is associated with tumorigenesis, inflammation,
and apoptosis. Small RNAs that modulate COX-2 gene expression would be useful
as tools to investigate biological functions of COX-2 in cells.
To investigate whether agRNAs have the potential to modulate COX-2 gene expression, duplex
RNAs targeting the promoter were designed and tested in A549 lung cancer cells for
gene activation. After transfecting agRNAs, western blot and qPCR analysis were performed
to check expression levels of COX-2 protein and mRNA. We found that some of the duplex
RNAs (agRNA-12 etc; 25 nM) can activate COX-2 gene expression by more than 20-fold
relative to mismatch controls. The fold activation was similar with that for IL-1b (10 ng/mL),
a natural activator for COX-2 gene. We observed more than 100-fold activation in COX-2
mRNA in cells treated with both the activating agRNA and IL-1b, which indicates the two
activators can work synergistically. ChIP analysis showed increased occupancy of RNA polymerase II by 3-5 fold and NFkB by 2-4 fold in the promoter, suggesting that this activation
occurs at the level of transcription. In our previous studies with PR and LDLR, we identified
non-coding transcripts overlapping their promoters, which could be potential targets for agRNAs.
We performed RACE and strand-specific RT-qPCR to examine whether similar noncoding
transcripts are expressed in the COX-2 gene promoter. Those analyses revealed that
sense and antinsense transcripts are expressed at the COX-2 gene promoter. These transcripts
might be an endogenous mediator for regulating COX-2 gene expression that can be targeted
by agRNAs. Transfection of activating agRNAs did not substantially change expression of interferon responsive genes such as OAS1, OAS2, MX1, IFITM1, and ISGF3g, suggesting that
the activation of COX-2 gene by agRNAs is not due to interferon response. These data suggest
that COX-2 gene can be induced by promoter-targeted duplex RNAs and the gene can beused as a model for research of RNA-mediated gene activation.
RNAi in humans: Phase I dose-escalation study of ALN-VSP02, a novel RNAi therapeutic for solid tumors with liver involvement
Background: ALN-VSP02 is a RNA interference (RNAi) therapeutic comprised of lipid
nanoparticle-formulated small interfering RNAs (siRNAs) targeting the expression of vascular
endothelial growth factor (VEGF)-A and kinesin spindle protein (KSP).
Methods: A multi-center, open label, Phase 1 dose-escalation trial of ALN-VSP02 administered
as a 15-minute iv infusion q2 wks was initiated in patients (pts) with advanced solid
tumors and at least one measurable liver lesion. Main objectives included evaluation of
safety/tolerability and assessment of PK/PD.
Results: Forty-one pts were enrolled across 7 dose levels (0.1-1.5 mg/kg); median age 57 yrs,
all with multiple prior therapies. A total of 182 doses have been administered to date, mean of
4.4 (range 1-24). Treatment was generally well-tolerated; the most common AEs were lowgrade
nausea, fatigue and fever with no clear dose-dependence. One on-study death (liver
failure in a pt with near complete replacement of the liver by tumor) deemed possibly related
to treatment occurred at 0.7 mg/kg. Grade 2 infusion-related reactions occurred in 15% of
pts or 3% of total doses administered and were managed with slowing of infusion. Grade 1-2
chills/rigors were seen in 3 of 11 pts at 1.25 mg/kg. Dose-limiting toxicities included reversible
grade 3 thrombocytopenia (2 pts at1.25 mg/kg) and hypokalemia (1 pt at 1.5 mg/kg).
Plasma PK showed dose-proportional AUC and Cmax. Post-treatment biopsies from 10 pts (7
liver and 3 extrahepatic tumors) showed pharmacologically relevant concentrations (2.2-142
ng/g tissue) of both siRNAs. Molecular evidence of RNAi-mediated VEGF mRNA cleavage
was shown in liver tumor biopsies (n=2 at 0.4 mg/kg) and in an extrahepatic tumor biopsy
(ovarian cancer at 1.25 mg/kg) through use of the 5’ RACE assay. Additional evidence for
an anti-VEGF effect with ALN-VSP02 included a decrease in Ktrans of at least 40% by
DCE-MRI in 46% of evaluable pts. Among 37 pts evaluable for response, 8% (1 of 13) at
doses ≤ 0.4 mg/kg had stable disease (SD) for at least 2 mo compared to 50% (12/24) with
SD (n=11) or PR (n=1, endometrial cancer with liver metastases) at doses ≥ 0.7 mg/kg.
Conclusions: ALN-VSP02 is well-tolerated and has antitumor activity. Pharmacodynamic
data are consistent with an anti-VEGF effect, and 1.0 mg/kg q2wks is the recommendedPhase II dose.
Regulus Presentations:
THERAPEUTIC TARG ETING OF microRNAs
Neil W. Gibson, Balkrishen Bhat, Christine C. Esau, Scott Davis, Jia Tay, Eric Marcusson,
Hubert Chen, Aimee L. Jackson, Lars Karlsson, 1Kathryn J. Moore, 2Andrei Goga
microRNAs act as master regulators in biological pathways, and are dysregulated in disease
areas including cancer, metabolism, fibrosis, and inflammation. Their ability to modulate
disease pathways makes targeting microRNAs an exciting new approach for drug discovery.
Oligonucleotides that inhibit microRNA function have been termed anti-miRs. Critical to the
development of anti-miRs as a therapeutic modality are chemical modifications to enhance
stability and target affinity, and an understanding of functional biodistribution of anti-miRs
to cells and tissues of therapeutic interest. Systemic and local delivery of unformulated antimiRs
enables broad distribution for targeting microRNA function in a diverse range of tissues
and cell types. We present recent advances in our use of anti-miRs against two specific
targets – miR-33 in metabolic disease and miR-21 in oncology.
miR-33a and b are intronic microRNAs located within the SREBF2 and SREBF1 genes,
respectively. This microRNA family suppresses the expression of the ABCA1 cholesterol
transporter and lowers HDL levels. LDL receptor-deficient mice treated with anti-miR-33
showed an increase in circulating HDL levels as well as enhanced reverse cholesterol
transport to the plasma, liver, and feces. The anti-miR-33-treated mice had reduced plaque
size and lipid content, increased markers of plaque stability, and decreased inflammatory gene
expression. The systemic delivery of an antisense oligonucleotide that targets both miR-33a
and miR-33b increases hepatic expression of ABCA1 and induces a sustained increase in
plasma HDL cholesterol in African green monkeys. These data suggest the modulation of
microRNA function as a promising strategy to treat atherosclerotic vascular disease.
miR-21 is frequently over-expressed and has been shown to correlate with poor outcome in
multiple cancer types. We have used a publically available data set from 86 patients to show
that miR-21 is over-expressed in hepatocellular carcinoma (HCC). Short term treatment with
the anti-miR-21 oligonucleotide in a genetically engineered mouse model of HCC led to a
reduction in tumor formation and an increased survival advantage. Furthermore, inhibition of
miR-21 was clearly demonstrated by analysis of genome wide mRNA expression data from
treated versus untreated tumors. Our findings suggest that miR-21 is a promising candidate
for the therapeutic intervention of liver cancer and further highlights the potential of antimiR-
mediated inhibition of microRNAs in cancer.
Overall our data suggest the therapeutic utility of anti-miRs targeting microRNAs involved inhuman disease pathogenesis.
THERAPEUTIC TARG ETING OF microRNAs
Neil W. Gibson, Balkrishen Bhat, Christine C. Esau, Scott Davis, Jia Tay, Eric Marcusson,
Hubert Chen, Aimee L. Jackson, Lars Karlsson, 1Kathryn J. Moore, 2Andrei Goga
microRNAs act as master regulators in biological pathways, and are dysregulated in disease
areas including cancer, metabolism, fibrosis, and inflammation. Their ability to modulate
disease pathways makes targeting microRNAs an exciting new approach for drug discovery.
Oligonucleotides that inhibit microRNA function have been termed anti-miRs. Critical to the
development of anti-miRs as a therapeutic modality are chemical modifications to enhance
stability and target affinity, and an understanding of functional biodistribution of anti-miRs
to cells and tissues of therapeutic interest. Systemic and local delivery of unformulated antimiRs
enables broad distribution for targeting microRNA function in a diverse range of tissues
and cell types. We present recent advances in our use of anti-miRs against two specific
targets – miR-33 in metabolic disease and miR-21 in oncology.
miR-33a and b are intronic microRNAs located within the SREBF2 and SREBF1 genes,
respectively. This microRNA family suppresses the expression of the ABCA1 cholesterol
transporter and lowers HDL levels. LDL receptor-deficient mice treated with anti-miR-33
showed an increase in circulating HDL levels as well as enhanced reverse cholesterol
transport to the plasma, liver, and feces. The anti-miR-33-treated mice had reduced plaque
size and lipid content, increased markers of plaque stability, and decreased inflammatory gene
expression. The systemic delivery of an antisense oligonucleotide that targets both miR-33a
and miR-33b increases hepatic expression of ABCA1 and induces a sustained increase in
plasma HDL cholesterol in African green monkeys. These data suggest the modulation of
microRNA function as a promising strategy to treat atherosclerotic vascular disease.
miR-21 is frequently over-expressed and has been shown to correlate with poor outcome in
multiple cancer types. We have used a publically available data set from 86 patients to show
that miR-21 is over-expressed in hepatocellular carcinoma (HCC). Short term treatment with
the anti-miR-21 oligonucleotide in a genetically engineered mouse model of HCC led to a
reduction in tumor formation and an increased survival advantage. Furthermore, inhibition of
miR-21 was clearly demonstrated by analysis of genome wide mRNA expression data from
treated versus untreated tumors. Our findings suggest that miR-21 is a promising candidate
for the therapeutic intervention of liver cancer and further highlights the potential of antimiR-
mediated inhibition of microRNAs in cancer.
Overall our data suggest the therapeutic utility of anti-miRs targeting microRNAs involved inhuman disease pathogenesis.
Tekmira Pharma presentations:
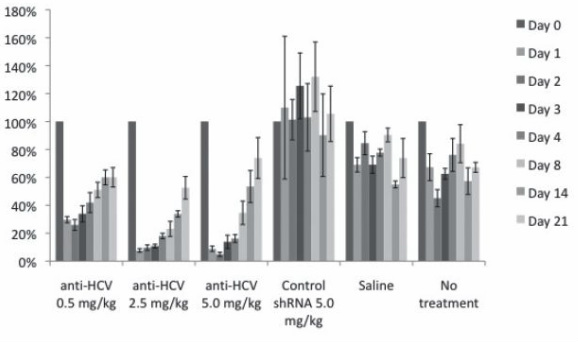
LIPID NANOPARTICLE FORMULATIONS OF MINIMAL-LENGTH shRNAS SHOW POTENT INHIBITION OF HCV-DRIVEN, LIVER-SPECIFIC GENE EXPRESSION IN MICE
B. H. Johnstona,e, A. Dallasa, H. Ilvesa, J. Shorensteina, M. A. Behlkeb, SP Wonge,
R. Harbottlec, and I. MacLachland
We have identified short shRNAs (sshRNAs) that target a highly conserved HCV sequence
and potently inhibit expression in reporter systems.1 Chemical modifications were subsequently
introduced to increase nuclease stability while minimizing immunostimulatory activity and
maintaining potency.2 In the current study, we assessed the efficiency of liver delivery and in
vivo efficacy of these sshRNAs when formulated into lipid nanoparticles (LNP). Single doses
of HCV-targeting sshRNAs formulated into LNP were injected i.v. into mice that had previously
been treated with a plasmid providing long-term expression of an HCV-firefly luciferase
fusion protein from a liver-specific promoter. In vivo imaging showed a dose-dependent inhibition
of luciferase expression (>90% at 2.5 mg/kg sshRNA), with a half-time of recovery of
about 3 weeks (see Figure; expression normalized to Day 0 = 100%). No inhibition was seen
with a scrambled control, saline, or a mock treatment. These results demonstrate the ability toprovide durable knockdown of an HCV target by systemic delivery of formulated sshRNAs.
LIPID NANOPARTICLE FORMULATIONS OF MINIMAL-LENGTH shRNAS SHOW POTENT INHIBITION OF HCV-DRIVEN, LIVER-SPECIFIC GENE EXPRESSION IN MICE
B. H. Johnstona,e, A. Dallasa, H. Ilvesa, J. Shorensteina, M. A. Behlkeb, SP Wonge,
R. Harbottlec, and I. MacLachland
We have identified short shRNAs (sshRNAs) that target a highly conserved HCV sequence
and potently inhibit expression in reporter systems.1 Chemical modifications were subsequently
introduced to increase nuclease stability while minimizing immunostimulatory activity and
maintaining potency.2 In the current study, we assessed the efficiency of liver delivery and in
vivo efficacy of these sshRNAs when formulated into lipid nanoparticles (LNP). Single doses
of HCV-targeting sshRNAs formulated into LNP were injected i.v. into mice that had previously
been treated with a plasmid providing long-term expression of an HCV-firefly luciferase
fusion protein from a liver-specific promoter. In vivo imaging showed a dose-dependent inhibition
of luciferase expression (>90% at 2.5 mg/kg sshRNA), with a half-time of recovery of
about 3 weeks (see Figure; expression normalized to Day 0 = 100%). No inhibition was seen
with a scrambled control, saline, or a mock treatment. These results demonstrate the ability toprovide durable knockdown of an HCV target by systemic delivery of formulated sshRNAs.
Competitor Presentations:
[Note that ISIS recently filed a patent infringement suit against Santaris, who licensed these programs to Enzon]
EVALUATION OF LOCKED NUCLEIC ACID (LNA)-BASED mRNA
ANTAGONI STS IN CANCER PATI ENTS
Buchbinder A, Kalambakas S, Huq N, Berkowitz N
Enzon Pharmaceuticals, Inc., Piscataway, NJ, USA
Background: HIF-1 is a transcription factor that is a critical mediator of angiogenesis, cell proliferation, metabolism, survival, and adaptive responses to stress. EZN-2968 is a potent locked nucleic acid antisense oligonucleotide that downmodulates HIF-1α mRNA and protein in
vitro (IC50 <1 nM and <5 nM, respectively). Survivin, the smallest member of the inhibitors of
apoptosis protein gene family, functions as a key regulator of mitosis and apoptosis. EZN-3042
is a potent locked nucleic acid antisense oligonucleotide that down-modulates survivin mRNA
and consequently protein in vitro (IC50 <5 nM). The LNA based mRNA antagonists EZN-2968
and EZN-3042 were evaluated in patients with cancer in Phase 1 clinical trials (1,2).
Methods: The primary objective of the studies was to determine the maximum tolerated doses
(MTD) and recommended Phase 2 dose. Secondary objectives were to evaluate safety, tolerability, pharmacokinetics (PK), and pharmacodynamics.
Results: EZN-2968: 49 pts received weekly EZN-2968 doses for 3, 4, or 5 of 6 weeks. Doselimiting toxicities (DLT) were intracerebral bleeding (n=1, 3.5 mg/kg), Grade 3 fatigue (n=2,
8 mg/kg, 18 mg/kg for 5 of 6 weeks), and Grade 3 increased AST (n=1, 18 mg/kg for 5 of 6
weeks). The MTD for EZN-2968 is 18 mg/kg given weekly for 4 out of 6 weeks. Drug-related
adverse events (AEs) reported for >10% of pts were fatigue (29%) and headache (14%). Grade
3 drug-related AEs (fatigue [n=3], AST increase [n=3], ALT increase [n=2], and hyponatremia
[n=1]) were reported for 7 patients (14%); no drug-related AE was Grade 4 or 5. The current
best response for EZN-2968 is stable disease (SD by RECIST) in 16 pts; 7 pts were treated
for >90 days. Objective tumor shrinkage was observed in several pts. HIF-1α expression was
evaluated pretreatment and on-treatment in tumor (n=6) and skin (n=41) samples. Tumor
biopsy HIF-1α mRNA decreased in 4 pts, increased in 1 pt, and did not change in 1 pt. Skin
biopsy HIF-1α mRNA decreased in 63% (26/41) of pts, remained the same in 10% (4/41) of
pts, and increased in 27% (11/41) of pts.EZN-3042: 24 pts received weekly EZN3042 doses for
4 out of 4 weeks. DLT was observed in 3 pts at 8 mg/kg (Grade 3 increased aspartate or alanine
aminotransferase [AST or ALT]). The MTD for EZN-3042 is 6.5 mg/kg. Drug-related adverse
events (AEs) (in >15% pts) were AST increase (42%), ALT increase (38%), fatigue (33%), and
diarrhea (17%). Most AEs were Grade 1 or 2. The best response was stable disease in 5 pts.
Conclusions: The MTD of EZN-2968 is 18 mg/kg given weekly for 4 out of 6 weeks. EZN-
2968 was generally well tolerated in previously treated pts with advanced tumors who received
up to 10 cycles of EZN-2968. The best response was SD. Evidence for down-regulation of the
HIF-1α target is supported by observations in tumor and skin biopsies. The MTD of EZN-3042is 6.5 mg/kg. EZN-3042 was well tolerated in previously treated pts with advanced tumors.
THE USE OF EXPRESSION PROFILES TO ASSESS NONHYBRIDISATION MEDIATED EFFECTS OF PHOSPHOROTHIOATE ANTISENSE OLIGONUCLEOTIDES
Peter H Hagedorn, Andreas Petri, Sakari Kauppinen, Morten Lindow
Santaris Pharma A/S, Kogle Alle 6, DK-2970 Hørsholm, Denmark
We have initiated systematic investigations of general class effects of LNA-modified phosphorothioate antisense oligonucleotides. To this end, we have created a reference set of
gene-expression profiles from cultured cells, rodents, and non-human primates treated with
different oligonucleotides, designated here as the Oligo Effect Map. Currently, this collection
comprises profiles from 28 LNA oligonucleotides contrasted to saline vehicle or mock transfection, and integrates more than 500 samples (in vivo from liver and kidney and in vitro from
cultured HuH-7, PC3, and HeLa cells) from 19 independent experiments. For each profile, we
have catalogued detailed experimental information (for example oligonucleotide sequence,
including positions with chemical modifications, delivery method, dosing regimen and dose
level/concentration used), as well as phenotypic observations for the animals representing the
profile. Using this resource we are able to detect gene expression patterns that occur in subsets
of experiments and correlate these to experimental setup and oligonucleotide design. Ultimately
this may provide us with useful information that enables the selection of conditions
(experimental or oligochemistry) avoid of unwanted effects, facilitating the design of efficientand safe RNA drugs.
THIRTEEN WEEK NON-CLINCIAL TESTING OF MIRAVIRSEN IN
CYNOMOLGUS MONKEYS
Hildebrandt-Eriksen L, Persson R, Foy J, Tessier Y, Levin AA
Santaris Pharma A/S, Hoersholm, Denmark
The safety and tolerability of miravirsen sodium (SPC3649), currently in phase 2 clinical
trials, was evaluated in a 13-week study with cynomolgus monkeys (Macaca fascicularis).
Miravirsen is an antagonist to microRNA-122 (miR-122), which in turn has been shown to be
a host factor in hepatitis C infection.
The subcutaneous dosing regimen was divided into a loading phase with 18, 40, 80 and 180
mg/kg/week respectively over 2 doses the first week and a maintenance phase with 2.5, 5, 10
and 25 mg/kg/week once weekly for 12 weeks followed by 12 weeks off treatment.
Miravirsen was well tolerated systemically and locally. Treatment-related changes in clinical
pathology parameters mainly consisted of decreased serum cholesterol level, an expected pharmacological
effect of miravirsen (as miR-122’s major function is the fine-tuning of lipid metabolism),
transient, mild aPTT and PT prolongation and activation of the alternative complement
pathway. Although some of these changes remained present through the 12-week treatment-free
period, they were considered not to be adverse owing to their small magnitude and/or since they
had no clinical or histopathological correlate. Microscopic findings were noted at all dose levels
and were considered to be related to the known class-related effects of oligonucleotides. The
kidneys were the main target organ. A dose-related accumulation of eosinophilic granules in the
proximal convoluted tubules associated with a minor level of degeneration accounted for some
functional disturbances in animals given ≥10 mg/kg/week and correlated with slightly increased
serum urea and creatinine levels. Changes in the kidney resolved at the end of the treatmentfree
period in most animals. The other histopathological effects (eg accumulation of enlarged
macrophages principally in the lymph nodes and liver (Kupffer cells) but also in injection
sites) were considered not to be adverse since they were not associated with any degenerative
changes. At the end of the treatment-free period, the vacuolated macrophages in the lymph
nodes showed a significant reduction in extent. Although some histopathological changes were
seen at 5 mg/kg/week, they were at a minimal level, in a few animals, and were not expected to
result in functional changes, including in the kidneys.
Liver and kidney samples were analyzed for miravirsen content after 13 weeks treatment and
12 weeks recovery, and in a small cohort of animals receiving 80mg/kg for the first week
only, on study days 5, 22, 43 and 70. The plasma pharmacokinetic evaluation confirmed the
long tissue half-life time of miravirsen, accounting for the incomplete reversibility of some of
the observed changes in tissues.The good safety profile of miravirsen sodium supports further clinical testing.
LNA ANTISENSE OLIGONUCLEOTIDES - A STRAIGHTFORWARD
CONCEPT FOR RNA THERAPEUTICS
Susanne Kammler¹, Niels Fisker Nielsen¹, Marie Lindholm¹, Robert E. Lanford², Andreas
Petri¹, Sakari Kauppinen¹, Nathalie Uzcategui¹, Ellen Marie Straarup¹, Joacim Elmén¹,
Troels Koch¹, Henrik Ørum¹, Maj Hedtjärn¹, Bo Rode Hansen¹
¹Santaris Pharma A/S, Hørsholm, Denmark
²Department of Virology and Immunology, Southwest Foundation for Biomedical Research,
San Antonio, TX 78227, USA
In the last decade, due to innovative oligonucleotide chemistry and designs, single stranded
antisense oligonucleotides have emerged as a highly promising therapeutic modality. Oligonucleotide mode of action spans from RNaseH mediated mRNA degradation to microRNA
inhibition and modulation of RNA splicing. Locked Nucleic Acid (LNA) oligonucleotides,
epitomise this new generation of effective RNA therapeutics, combining high binding affinity,
target specificity, regulatory potency and nuclease stability with unmatched short length of
the antisense oligonucleotide. It has been shown that LNA oligonucleotides readily enter cells
via un-assisted uptake (gymnosis), in an active form allowing in vitro prediction of potent
therapeutically active compounds as well as in vivo distribution to manifold cell types and tissues.
This makes Santaris Pharma A/S’ Locked Nucleic Acid (LNA) drug discovery platform
a straightforward tool to transform today’s knowledge about disease associated gene regulation into tomorrow’s RNA therapeutics.
Are short oligonucleotides less specific in vivo?
Morten Lindow, Peter Hagedorn, Andreas Petri
Santaris Pharma A/S, Kogle Alle 6, DK-2970 Hørsholm, Denmark
The challenge of developing effective and safe RNA-targeted therapeutics calls for robust in
silico design of target-specific oligonucleotides with minimal effect on unintended targets. In
this process, many molecular biologists and statisticians often ask how many perfect match
recognition sequencesa given oligo-nucleotide has in the transcriptome. This, in turn, leads to
the assumption that the longer a given oligonucleotide, the more specific it will be, whereas
short oligonucleotides are often considered unspecific and to have more off-target effects.
However, this assumptionis only correct if the hybridization stringency can be controlled in
such a way that a single mismatch, an insertion, or a deletion will abrogate effective binding
to unintendedtargets.This is indeed the case in most experiments such as PCR, Northern blot
analysis, and in situ hybridization. The primary goal is to find experimental settings in which
the oligonucleotidepreferably hybridizes to the intended target. In the lab, hybridization
conditions such as temperature and ionic strength can be controlled to achieve optimal specificity.
By comparison, hybridization conditionsin live animals or humans cannot be adjusted,
instead altering the length, design and chemical modifications of a given oligonucleotide
must be utilized to achieve adequate specificity.
Here, we present and compare different oligonucleotide specificity prediction algorithms.
We show that different methods can produce qualitatively very different results. Algorithms
based on minimizing mismatches predict that short oligonucleotides are less specific, whereas
methods that maximize the number of basepairs or binding affinity to putative targets predict
that longer oligonucleotides are less specific.We assess the aforementioned approaches using
transcriptomal expression profiling of mice livers after treatment with different LNA phosphorothioate oligonucleotides.
SILENCING OF microRNA FAMILIES BY SEED-TARGETING
TINY LNAs
Susanna Obad1, Andreas Petri1, Oliver Broom1, Markus Heidenblad1, Jan Stenvang1,
Ellen Marie Straarup1, Troels Koch1, Henrik Frydenlund Hansen1 and Sakari Kauppinen1,2
1Santaris Pharma, Kogle Allé 6, DK-2970 Hørsholm, Denmark
2Copenhagen Institute of Technology, Aalborg University, Lautrupvang 15, DK-2750
Ballerup, Denmark
MicroRNAs act as important post-transcriptional regulators of gene expression by mediating
mRNA degradation or translational repression. There is now ample evidence that perturbations
in the levels of individual or entire families of miRNAs are prevalent in and strongly
associated with the development of a variety of human diseases. Apart from cancer, miRNAs
have also been implicated in viral infections, cardiovascular disease and neurological disorders.
Hence, disease-associated miRNAs could represent a potential new class of targets for
oligonucleotide-based therapeutics, which may yield patient benefits unobtainable by other
therapeutic approaches.
LNAs comprise a class of bicyclic high-affinity RNA analogues, in which the ribose ring
in the sugar-phosphate backbone is locked in an RNA-like, C3’-endo conformation by the
introduction of a 2’-O,4’-C methylene bridge. This results in high binding affinity of singlestranded
LNA-modified oligonucleotides to their complementary miRNA targets. Moreover,
LNAs combined with a phosphorothioate backbone show high biostability and enhanced
pharmacokinetic properties in vivo. Here, we describe an approach that enables miRNA
knock-down using 8 nucleotide fully LNA-modified phosphorothioate oligonucleotides,
termed tiny LNAs, complementary to the miRNA seed region. Transfection of tiny LNAs
into cells resulted in simultaneous inhibition of miRNAs within families sharing the same
seed with concomitant up-regulation of direct targets. In addition, systemically delivered,
unconjugated tiny LNAs showed uptake in many normal tissues, and pharmacological effect
in different mouse models, coinciding with long-term miRNA silencing. Considered together,
these data support the utility of tiny LNAs in elucidating the functions of miRNA families
with important implications for the development of therapeutic strategies aiming at pharmacologicalinhibition of disease-associated miRNAs.
PLASMA PHARMACOKINETICS OF MIRAVIRSEN IN HUMANS
Persson R, Bagger Y, Børgesen H, Hodges, MR, King BD, Levin AA
Santaris Pharma A/S, Hørsholm, DK, San Diego CA USA
MiR-122, an abundant hepatic microRNA is an obligate host factor for HCV propagation and
represents a unique target for Hepatitis C Virus (HCV) therapy. The pharmacokinetics of a
LNA-modified antisense oligonucleotide complementary to miR-122, (miravirsen sodium),
was studied after single or multiple doses (i.v.) or (s.c.) to healthy subjects. Subjects were
divided into 6 groups (receiving 5 doses between 1.0 and 5.0 mg/kg/week). Each group
consisted of 5 individuals (4 active and 1 placebo). Plasma concentration of miravirsen was
quantitated using a validated hybridization-dependent ELISA. Plasma pharmacokinetics
(e.g. AUC values) did not substantially change moving from a single dose to multiple doses
and exposures after i.v. and s.c. administrations were similar. The plasma PK for miravirsen
were multiphasic with distribution phase(s) and a terminal phase. The distribution phase was
completed within 24 hours (t½≈1-2 hours) and the plasma compartment was almost freed of
miravirsen (to > 99%). The terminal phase lasted for weeks (t½=38 days). Comparison of
AUCs following s.c. and i.v. doses indicated bioavailability was 100%. The terminal PK
parameters (t½, Vz and Cl) were similar independent of route. AUC0-96 h and Cmax after s.c.
administration increased dose –proportionally for AUC0-96 h, and less than dose-proportionally
for Cmax on both Days 1 and 29. The mean ratio of AUCday29/AUCday1 was 1.0, indicating
that there was no accumulation after 5 doses. Terminal PK parameters were estimated from
extended sampling (up to 140 days) after the last dose on Day 29. Total body clearance, Cl/F,
was low and independent of dose, with a mean value of 36.0 (± 8.5) mL/h/kg (all s.c. doses
combined). The apparent volume of distribution, Vz/F, was large and independent of dose,
with a mean value of 45.4 (± 14) L/kg, indicating a distribution into a deeper compartment
e.g. liver and kidney. Mean terminal plasma half-life was 38.3 (± 16) days (all s.c. dose levels
combined). While there was no significant accumulation of miravirsen in plasma, equilibrium
or trough plasma concentrations increased linearly with dose and with number of doses,
reflecting accumulation in deeper compartments. These data will be used to design dosingschedules for future clinical trials.
[Note that ISIS recently filed a patent infringement suit against Santaris, who licensed these programs to Enzon]
EVALUATION OF LOCKED NUCLEIC ACID (LNA)-BASED mRNA
ANTAGONI STS IN CANCER PATI ENTS
Buchbinder A, Kalambakas S, Huq N, Berkowitz N
Enzon Pharmaceuticals, Inc., Piscataway, NJ, USA
Background: HIF-1 is a transcription factor that is a critical mediator of angiogenesis, cell proliferation, metabolism, survival, and adaptive responses to stress. EZN-2968 is a potent locked nucleic acid antisense oligonucleotide that downmodulates HIF-1α mRNA and protein in
vitro (IC50 <1 nM and <5 nM, respectively). Survivin, the smallest member of the inhibitors of
apoptosis protein gene family, functions as a key regulator of mitosis and apoptosis. EZN-3042
is a potent locked nucleic acid antisense oligonucleotide that down-modulates survivin mRNA
and consequently protein in vitro (IC50 <5 nM). The LNA based mRNA antagonists EZN-2968
and EZN-3042 were evaluated in patients with cancer in Phase 1 clinical trials (1,2).
Methods: The primary objective of the studies was to determine the maximum tolerated doses
(MTD) and recommended Phase 2 dose. Secondary objectives were to evaluate safety, tolerability, pharmacokinetics (PK), and pharmacodynamics.
Results: EZN-2968: 49 pts received weekly EZN-2968 doses for 3, 4, or 5 of 6 weeks. Doselimiting toxicities (DLT) were intracerebral bleeding (n=1, 3.5 mg/kg), Grade 3 fatigue (n=2,
8 mg/kg, 18 mg/kg for 5 of 6 weeks), and Grade 3 increased AST (n=1, 18 mg/kg for 5 of 6
weeks). The MTD for EZN-2968 is 18 mg/kg given weekly for 4 out of 6 weeks. Drug-related
adverse events (AEs) reported for >10% of pts were fatigue (29%) and headache (14%). Grade
3 drug-related AEs (fatigue [n=3], AST increase [n=3], ALT increase [n=2], and hyponatremia
[n=1]) were reported for 7 patients (14%); no drug-related AE was Grade 4 or 5. The current
best response for EZN-2968 is stable disease (SD by RECIST) in 16 pts; 7 pts were treated
for >90 days. Objective tumor shrinkage was observed in several pts. HIF-1α expression was
evaluated pretreatment and on-treatment in tumor (n=6) and skin (n=41) samples. Tumor
biopsy HIF-1α mRNA decreased in 4 pts, increased in 1 pt, and did not change in 1 pt. Skin
biopsy HIF-1α mRNA decreased in 63% (26/41) of pts, remained the same in 10% (4/41) of
pts, and increased in 27% (11/41) of pts.EZN-3042: 24 pts received weekly EZN3042 doses for
4 out of 4 weeks. DLT was observed in 3 pts at 8 mg/kg (Grade 3 increased aspartate or alanine
aminotransferase [AST or ALT]). The MTD for EZN-3042 is 6.5 mg/kg. Drug-related adverse
events (AEs) (in >15% pts) were AST increase (42%), ALT increase (38%), fatigue (33%), and
diarrhea (17%). Most AEs were Grade 1 or 2. The best response was stable disease in 5 pts.
Conclusions: The MTD of EZN-2968 is 18 mg/kg given weekly for 4 out of 6 weeks. EZN-
2968 was generally well tolerated in previously treated pts with advanced tumors who received
up to 10 cycles of EZN-2968. The best response was SD. Evidence for down-regulation of the
HIF-1α target is supported by observations in tumor and skin biopsies. The MTD of EZN-3042is 6.5 mg/kg. EZN-3042 was well tolerated in previously treated pts with advanced tumors.
THE USE OF EXPRESSION PROFILES TO ASSESS NONHYBRIDISATION MEDIATED EFFECTS OF PHOSPHOROTHIOATE ANTISENSE OLIGONUCLEOTIDES
Peter H Hagedorn, Andreas Petri, Sakari Kauppinen, Morten Lindow
Santaris Pharma A/S, Kogle Alle 6, DK-2970 Hørsholm, Denmark
We have initiated systematic investigations of general class effects of LNA-modified phosphorothioate antisense oligonucleotides. To this end, we have created a reference set of
gene-expression profiles from cultured cells, rodents, and non-human primates treated with
different oligonucleotides, designated here as the Oligo Effect Map. Currently, this collection
comprises profiles from 28 LNA oligonucleotides contrasted to saline vehicle or mock transfection, and integrates more than 500 samples (in vivo from liver and kidney and in vitro from
cultured HuH-7, PC3, and HeLa cells) from 19 independent experiments. For each profile, we
have catalogued detailed experimental information (for example oligonucleotide sequence,
including positions with chemical modifications, delivery method, dosing regimen and dose
level/concentration used), as well as phenotypic observations for the animals representing the
profile. Using this resource we are able to detect gene expression patterns that occur in subsets
of experiments and correlate these to experimental setup and oligonucleotide design. Ultimately
this may provide us with useful information that enables the selection of conditions
(experimental or oligochemistry) avoid of unwanted effects, facilitating the design of efficientand safe RNA drugs.
THIRTEEN WEEK NON-CLINCIAL TESTING OF MIRAVIRSEN IN
CYNOMOLGUS MONKEYS
Hildebrandt-Eriksen L, Persson R, Foy J, Tessier Y, Levin AA
Santaris Pharma A/S, Hoersholm, Denmark
The safety and tolerability of miravirsen sodium (SPC3649), currently in phase 2 clinical
trials, was evaluated in a 13-week study with cynomolgus monkeys (Macaca fascicularis).
Miravirsen is an antagonist to microRNA-122 (miR-122), which in turn has been shown to be
a host factor in hepatitis C infection.
The subcutaneous dosing regimen was divided into a loading phase with 18, 40, 80 and 180
mg/kg/week respectively over 2 doses the first week and a maintenance phase with 2.5, 5, 10
and 25 mg/kg/week once weekly for 12 weeks followed by 12 weeks off treatment.
Miravirsen was well tolerated systemically and locally. Treatment-related changes in clinical
pathology parameters mainly consisted of decreased serum cholesterol level, an expected pharmacological
effect of miravirsen (as miR-122’s major function is the fine-tuning of lipid metabolism),
transient, mild aPTT and PT prolongation and activation of the alternative complement
pathway. Although some of these changes remained present through the 12-week treatment-free
period, they were considered not to be adverse owing to their small magnitude and/or since they
had no clinical or histopathological correlate. Microscopic findings were noted at all dose levels
and were considered to be related to the known class-related effects of oligonucleotides. The
kidneys were the main target organ. A dose-related accumulation of eosinophilic granules in the
proximal convoluted tubules associated with a minor level of degeneration accounted for some
functional disturbances in animals given ≥10 mg/kg/week and correlated with slightly increased
serum urea and creatinine levels. Changes in the kidney resolved at the end of the treatmentfree
period in most animals. The other histopathological effects (eg accumulation of enlarged
macrophages principally in the lymph nodes and liver (Kupffer cells) but also in injection
sites) were considered not to be adverse since they were not associated with any degenerative
changes. At the end of the treatment-free period, the vacuolated macrophages in the lymph
nodes showed a significant reduction in extent. Although some histopathological changes were
seen at 5 mg/kg/week, they were at a minimal level, in a few animals, and were not expected to
result in functional changes, including in the kidneys.
Liver and kidney samples were analyzed for miravirsen content after 13 weeks treatment and
12 weeks recovery, and in a small cohort of animals receiving 80mg/kg for the first week
only, on study days 5, 22, 43 and 70. The plasma pharmacokinetic evaluation confirmed the
long tissue half-life time of miravirsen, accounting for the incomplete reversibility of some of
the observed changes in tissues.The good safety profile of miravirsen sodium supports further clinical testing.
LNA ANTISENSE OLIGONUCLEOTIDES - A STRAIGHTFORWARD
CONCEPT FOR RNA THERAPEUTICS
Susanne Kammler¹, Niels Fisker Nielsen¹, Marie Lindholm¹, Robert E. Lanford², Andreas
Petri¹, Sakari Kauppinen¹, Nathalie Uzcategui¹, Ellen Marie Straarup¹, Joacim Elmén¹,
Troels Koch¹, Henrik Ørum¹, Maj Hedtjärn¹, Bo Rode Hansen¹
¹Santaris Pharma A/S, Hørsholm, Denmark
²Department of Virology and Immunology, Southwest Foundation for Biomedical Research,
San Antonio, TX 78227, USA
In the last decade, due to innovative oligonucleotide chemistry and designs, single stranded
antisense oligonucleotides have emerged as a highly promising therapeutic modality. Oligonucleotide mode of action spans from RNaseH mediated mRNA degradation to microRNA
inhibition and modulation of RNA splicing. Locked Nucleic Acid (LNA) oligonucleotides,
epitomise this new generation of effective RNA therapeutics, combining high binding affinity,
target specificity, regulatory potency and nuclease stability with unmatched short length of
the antisense oligonucleotide. It has been shown that LNA oligonucleotides readily enter cells
via un-assisted uptake (gymnosis), in an active form allowing in vitro prediction of potent
therapeutically active compounds as well as in vivo distribution to manifold cell types and tissues.
This makes Santaris Pharma A/S’ Locked Nucleic Acid (LNA) drug discovery platform
a straightforward tool to transform today’s knowledge about disease associated gene regulation into tomorrow’s RNA therapeutics.
Are short oligonucleotides less specific in vivo?
Morten Lindow, Peter Hagedorn, Andreas Petri
Santaris Pharma A/S, Kogle Alle 6, DK-2970 Hørsholm, Denmark
The challenge of developing effective and safe RNA-targeted therapeutics calls for robust in
silico design of target-specific oligonucleotides with minimal effect on unintended targets. In
this process, many molecular biologists and statisticians often ask how many perfect match
recognition sequencesa given oligo-nucleotide has in the transcriptome. This, in turn, leads to
the assumption that the longer a given oligonucleotide, the more specific it will be, whereas
short oligonucleotides are often considered unspecific and to have more off-target effects.
However, this assumptionis only correct if the hybridization stringency can be controlled in
such a way that a single mismatch, an insertion, or a deletion will abrogate effective binding
to unintendedtargets.This is indeed the case in most experiments such as PCR, Northern blot
analysis, and in situ hybridization. The primary goal is to find experimental settings in which
the oligonucleotidepreferably hybridizes to the intended target. In the lab, hybridization
conditions such as temperature and ionic strength can be controlled to achieve optimal specificity.
By comparison, hybridization conditionsin live animals or humans cannot be adjusted,
instead altering the length, design and chemical modifications of a given oligonucleotide
must be utilized to achieve adequate specificity.
Here, we present and compare different oligonucleotide specificity prediction algorithms.
We show that different methods can produce qualitatively very different results. Algorithms
based on minimizing mismatches predict that short oligonucleotides are less specific, whereas
methods that maximize the number of basepairs or binding affinity to putative targets predict
that longer oligonucleotides are less specific.We assess the aforementioned approaches using
transcriptomal expression profiling of mice livers after treatment with different LNA phosphorothioate oligonucleotides.
SILENCING OF microRNA FAMILIES BY SEED-TARGETING
TINY LNAs
Susanna Obad1, Andreas Petri1, Oliver Broom1, Markus Heidenblad1, Jan Stenvang1,
Ellen Marie Straarup1, Troels Koch1, Henrik Frydenlund Hansen1 and Sakari Kauppinen1,2
1Santaris Pharma, Kogle Allé 6, DK-2970 Hørsholm, Denmark
2Copenhagen Institute of Technology, Aalborg University, Lautrupvang 15, DK-2750
Ballerup, Denmark
MicroRNAs act as important post-transcriptional regulators of gene expression by mediating
mRNA degradation or translational repression. There is now ample evidence that perturbations
in the levels of individual or entire families of miRNAs are prevalent in and strongly
associated with the development of a variety of human diseases. Apart from cancer, miRNAs
have also been implicated in viral infections, cardiovascular disease and neurological disorders.
Hence, disease-associated miRNAs could represent a potential new class of targets for
oligonucleotide-based therapeutics, which may yield patient benefits unobtainable by other
therapeutic approaches.
LNAs comprise a class of bicyclic high-affinity RNA analogues, in which the ribose ring
in the sugar-phosphate backbone is locked in an RNA-like, C3’-endo conformation by the
introduction of a 2’-O,4’-C methylene bridge. This results in high binding affinity of singlestranded
LNA-modified oligonucleotides to their complementary miRNA targets. Moreover,
LNAs combined with a phosphorothioate backbone show high biostability and enhanced
pharmacokinetic properties in vivo. Here, we describe an approach that enables miRNA
knock-down using 8 nucleotide fully LNA-modified phosphorothioate oligonucleotides,
termed tiny LNAs, complementary to the miRNA seed region. Transfection of tiny LNAs
into cells resulted in simultaneous inhibition of miRNAs within families sharing the same
seed with concomitant up-regulation of direct targets. In addition, systemically delivered,
unconjugated tiny LNAs showed uptake in many normal tissues, and pharmacological effect
in different mouse models, coinciding with long-term miRNA silencing. Considered together,
these data support the utility of tiny LNAs in elucidating the functions of miRNA families
with important implications for the development of therapeutic strategies aiming at pharmacologicalinhibition of disease-associated miRNAs.
PLASMA PHARMACOKINETICS OF MIRAVIRSEN IN HUMANS
Persson R, Bagger Y, Børgesen H, Hodges, MR, King BD, Levin AA
Santaris Pharma A/S, Hørsholm, DK, San Diego CA USA
MiR-122, an abundant hepatic microRNA is an obligate host factor for HCV propagation and
represents a unique target for Hepatitis C Virus (HCV) therapy. The pharmacokinetics of a
LNA-modified antisense oligonucleotide complementary to miR-122, (miravirsen sodium),
was studied after single or multiple doses (i.v.) or (s.c.) to healthy subjects. Subjects were
divided into 6 groups (receiving 5 doses between 1.0 and 5.0 mg/kg/week). Each group
consisted of 5 individuals (4 active and 1 placebo). Plasma concentration of miravirsen was
quantitated using a validated hybridization-dependent ELISA. Plasma pharmacokinetics
(e.g. AUC values) did not substantially change moving from a single dose to multiple doses
and exposures after i.v. and s.c. administrations were similar. The plasma PK for miravirsen
were multiphasic with distribution phase(s) and a terminal phase. The distribution phase was
completed within 24 hours (t½≈1-2 hours) and the plasma compartment was almost freed of
miravirsen (to > 99%). The terminal phase lasted for weeks (t½=38 days). Comparison of
AUCs following s.c. and i.v. doses indicated bioavailability was 100%. The terminal PK
parameters (t½, Vz and Cl) were similar independent of route. AUC0-96 h and Cmax after s.c.
administration increased dose –proportionally for AUC0-96 h, and less than dose-proportionally
for Cmax on both Days 1 and 29. The mean ratio of AUCday29/AUCday1 was 1.0, indicating
that there was no accumulation after 5 doses. Terminal PK parameters were estimated from
extended sampling (up to 140 days) after the last dose on Day 29. Total body clearance, Cl/F,
was low and independent of dose, with a mean value of 36.0 (± 8.5) mL/h/kg (all s.c. doses
combined). The apparent volume of distribution, Vz/F, was large and independent of dose,
with a mean value of 45.4 (± 14) L/kg, indicating a distribution into a deeper compartment
e.g. liver and kidney. Mean terminal plasma half-life was 38.3 (± 16) days (all s.c. dose levels
combined). While there was no significant accumulation of miravirsen in plasma, equilibrium
or trough plasma concentrations increased linearly with dose and with number of doses,
reflecting accumulation in deeper compartments. These data will be used to design dosingschedules for future clinical trials.
 RSS Feed
RSS Feed